A series of device malfunctions, coupled with technological and scientific advances in the medical device industry, drove regulators to revamp the decades-old European medical device regulations (the Medical Device Directive or MDD and the In Vitro Diagnostic Directive or IVDD). The European Commission introduced the European Medical Device Regulation (EU MDR) and the In Vitro Diagnostic Regulation (IVDR) on May 26, 2017, with the actual launch expected in May 2021 and May 2022 respectively. The primary goal was to strengthen the safety and effectiveness of medical devices commercialized in the European market. EU MDR was earlier set for launch in May 2020, however, a series of overwhelming events – primarily the global COVID-19 pandemic, along with the industry’s under-preparedness to comply – has forced EU regulators to consider a one-year implementation delay.
The new regulations put stringent controls on device classification, clinical evaluation, and post-market surveillance and are expected to have unique implications for all stakeholders across the industry. To achieve EU MDR and IVDR compliance, we recommend medical device manufacturers to take an organization-wide approach with C-suite involvement and redefine their compliance strategies to remain relevant in the European market.
Scope
Industry: medical devices
Geography: European Union
Contents
In this viewpoint, we discuss the following topics:
An overview of EU MDR & IVDR and their objectives
Challenges in implementing the new regulations
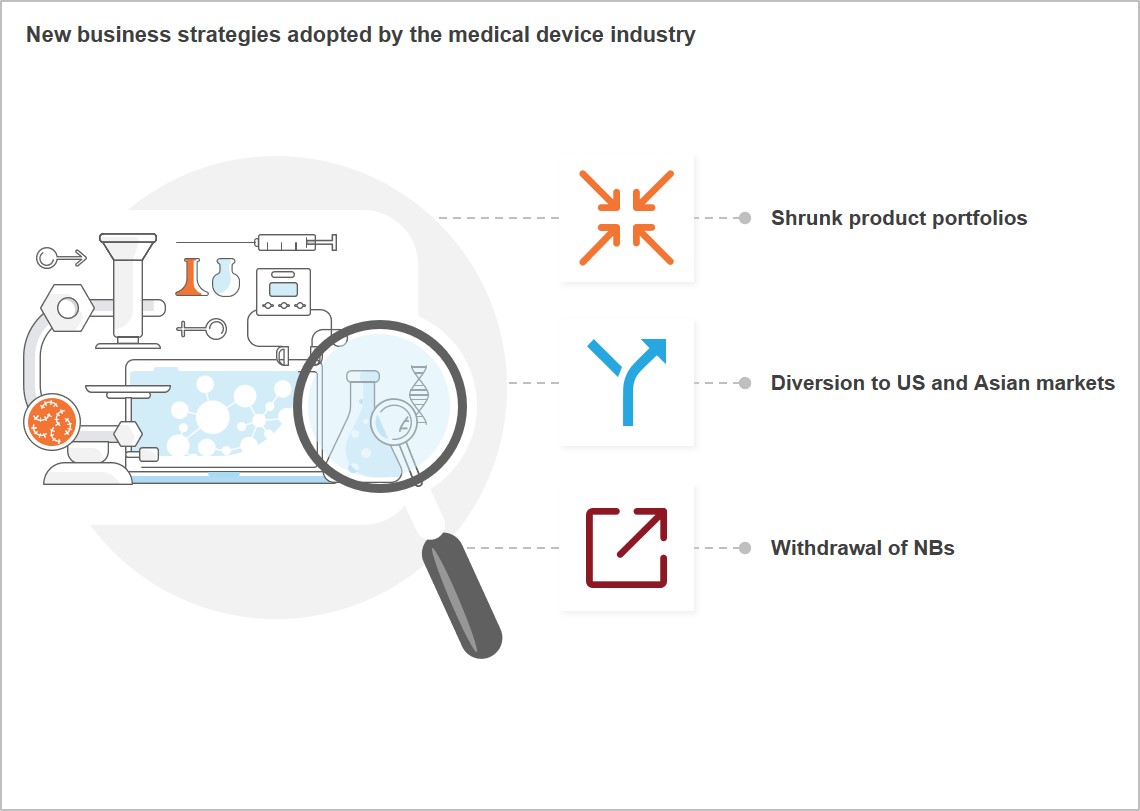
The impact of EU MDR & IVDR on the medical device industry
How are medical device manufacturers, CROs, and services providers likely to react to these new regulations?
What are the steps being taken by players across the ecosystem (to ensure compliance with the new regulations?